只有高质量的研究设计才是研发抗癌新药造福于全球癌症患者的基石
2018-06-17 慧语 贾老师 肿瘤资讯 良医汇
肿瘤药物研发日趋成熟,无论是制药行业还是政府监管部门亦或是其它学术组织,都迫切希望在评价抗癌新药、尤其是“变革性”创新药疗效时能尽可能地提高效率、降低成本,加速满足癌症患者的迫切需求。但提速的同时兼顾时效性和伦理学问题仍不容忽视。在时代的机遇和使命面前,无缝(Seamless)设计作为一种创新性的临床试验理念能否担此重任,脱颖而出呢?--请看新英格兰杂志如何评价。
肿瘤药物研发日趋成熟,无论是制药行业还是政府监管部门亦或是其它学术组织,都迫切希望在评价抗癌新药、尤其是“变革性”创新药疗效时能尽可能地提高效率、降低成本,加速满足癌症患者的迫切需求。但提速的同时兼顾时效性和伦理学问题仍不容忽视。在时代的机遇和使命面前,无缝(Seamless)设计作为一种创新性的临床试验理念能否担此重任,脱颖而出呢?--请看新英格兰杂志如何评价。

半个多世纪以来,抗癌药物的开发都遵循一个传统的模式,即I期临床试验确定药物的安全性、耐受性和剂量;II期临床试验初步评价药物对各种癌症患者的治疗作用和安全性;III期临床试验主要是进一步确证新药相对于现有方法的疗效和安全性,是监管部门批准的主要依据。在过去十年中对癌症生物学了解的进步,促进了更为有效药物的开放以及根据生物标志物进行早期诊断,为患者提供了更多有效的治疗方案。但由于癌症的复杂性,对于大多数患者而言仍是不治之症,因此患者对于能够尝试使用具有“变革性”的创新抗癌药物治疗具有强烈的需求。而制药企业和监管层也希望能够加快抗癌新药的开发和批准过程,造福广大患者。由此,原本三个阶段的药物开发也因此变得越来越模糊。
为抢占市场先机,各大药厂纷纷启动无缝设计研究
2011年默沙东开展的一项I期临床试验(KEYNOTE-001,NCT01295827),以确定帕博利珠单抗(pembrolizumab)治疗晚期实体瘤患者的安全性和推荐剂量。受试患者的缓解率和持续缓解时间在试验早期就有非常显著的提升,尤其是对于转移性黑色素瘤和非小细胞肺癌患者疗效格外好。但为评估疗效和不同的给药方案以及挖掘潜在生物标志物,需要增加其他队列,导致最终额外纳入1200多名患者。本试验开始仅3年后,仅依据KEYNOTE-001研究其中一个173例患者队列的主要结果作为证据,帕博利珠单抗通过加速审批通道获批治疗黑素瘤患者。KEYNOTE-001研究后续的数据还被用来支持非小细胞肺癌的加速审批,以及对应诊断试剂的获批。
鉴于帕博利珠单抗开发过程超高的效率以及最终在一些适应症上成功获批,许多医药公司已经放弃了传统的三阶段的药物开发流程,转而选择在I期临床试验中采取无缝(seamless)设计的方法,评价药物在各癌种的活性和适当的剂量。目前基于无缝设计商业开发的获批免疫药物见表1。
表1 基于无缝(Seamless)设计获批的免疫药物
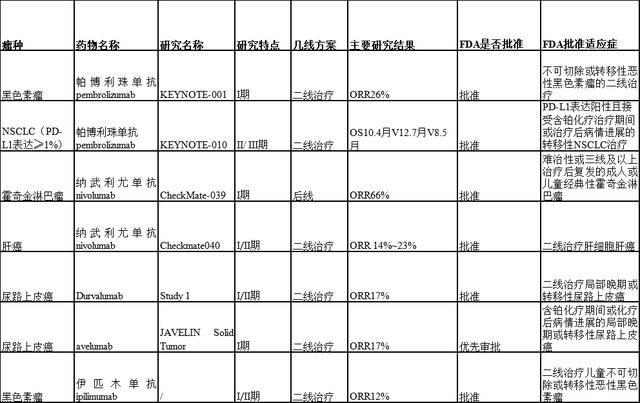
尽管帕博利珠单抗的例子诠释了一种高效药物开发——“无缝药物开发”模式,但包括临床研究者、审查机构、生物统计学家、制药公司、患者和监管者在内的,多个支持重大创新抗癌药物的快速开发和审批的团体,都表达了对I期临床试验中迅速扩大样本量的担忧。
关于无缝设计曾公开探讨过一些共同关注的问题,主要涉及一些在传统分期药物开发中公认,但在扩展队列设计试验中缺失的标准元素。例如,所有的临床试验都应该有明确的目标,以及具有能够实现这些目标的设计方案和统计分析计划。对于预计加入多个扩展队列的试验,这意味着需要前瞻性地确定扩展队列的定义、数量和样本量计算的理论基础。这也意味着研究中需要明确扩展队列患者的入选标准,并接受审查委员会及监察机构的审核。并且对于需要加入扩展队列的研究,应注意知情同意书的及时更新:新扩展队列的受试者在签署知情同意书时,应当详细告知患者药物的最新安全性和重要疗效评价数据。
在I期临床试验中加入额外的扩展队列,可能会出现一个临床试验就包括一个完整药物开发的情况,原本在传统分期临床研究中,各期之间潜在的重要保护作用也因此可能会消失。例如在II期试验结束与III期试验启动之间召开的申办方和FDA之间的标准指导会议,讨论后期药物开发需要注重和改善的问题,但此环节则将这种“无缝”设计研究中被省略。
此外,对于具有多个癌种的扩展队列试验,本应由FDA进行监管监督的疗效评价,可能最终会由一个专门的肿瘤学团队进行评估,而后者对药物的审批上市并无监管责任。虽然在最终的药物获批时可获得来自多个队列的更大的安全性数据集,但由于研究过程中缺乏对照组,可能难以全面评估药物的毒性。
最后,来自单个治疗组的试验数据可能不足以支持在一些国家通过监管审批,该药物在全球范围内使用的计划可能被推迟或限制
鼓励“无缝药物研发”的同时也应注重保护患者利益
由于无缝扩展队列设计能够大幅提升药物开发效率,但同时此类试验也具有一定的风险,因此必须为参加这种试验的患者提供一些保障措施。我们建议FDA的“突破疗法”通道,用来审批那些早期疗效证据足以证明该药物适合进行一个无缝研究的药物。只有那些早期临床证据表明新药较现有药物疗效具有显著改善时,才可以通过“突破疗法”通道。这种分配方式将确保在整个药物开发过程中的高标准监督,并且申办方与FDA涉及的多学科之间进行更为频繁、密集、及时的交流。限制“突破疗法”审批中使用无缝设计的研究,能够解决许多关于使用这种药物开发模式所引发的问题。
此外,在无缝设计的试验中使用独立的数据和安全监测委员会,也是保证数据质量的关键。一项临床试验需要独立监管委员会必要的监督不是一个新概念,也不是一个沉重的负担。在一个多扩展队列的试验中,独立监察委员会将按照预定的时间来审查现有队列中患者的安全性和有效性数据,建议研究人员增加或关闭队列,同时将整个研究的结果公开、透明,避免暗箱操作,确保试验的统计完整性。
针对具有前所未有的“颠覆性”抗癌药物(如免疫抗癌药物)的研发过程中,应鼓励开展无缝扩展队列设计的临床试验,达到早日“治病救人”的目的。而传统分期试验设计中为受试者提供的保护,可以通过更仔细地筛选适合此类设计方案的药物、更关注统计分析的理论基础和额外队列的分析计划、建立外部监督委员会、更频繁实时地在申办方、调查员、IRB、监管者和患者之间进行交流等措施,更好地保护患者权益。上述讨论关注和潜在的解决方案也是美国癌症研究协会年会关于监管科学和政策的会议的主题,具体细节将在FDA血液和肿瘤学办公室目前正在起草的行业准则中进一步阐述。
结语
为患者尽早提供更为有效的抗癌药物是一贯努力的方向,事实上,各大制药企业近年来采用的无缝研发模式的确是给癌症患者带来了更多的治疗选择和获益。但我们不能因此放弃曾经对开展临床试验的承诺——“精心设计、缜密执行”。只有来自高质量研究设计的数据,才会使医生在临床治疗中充分了解药物的风险和获益,从而保证患者的利益。也只有高质量的研究设计才是研发卓越抗癌新药造福于全球癌症患者的基石!
原始出处:
Prowell T M, Theoret M R, Pazdur R. Seamless Oncology-Drug Development[J]. New England Journal of Medicine, 2016, 374(21):2001.
作者:慧语 贾老师
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言
小提示:本篇资讯需要登录阅读,点击跳转登录





#抗癌新药#
51
学习了.长知识
81
#癌症患者#
32
#研发#
42
学习了受益匪浅
85