2岁多孩子已2000多度近视
某孩子一生下来就是个大胖小子:身长55厘米,体重4.45公斤,块头比普通孩子大许多。但他的成长之路并不顺利,9个月大的时候,家人就发现他看电视总往前凑,看东西似乎与别的孩子不一样。
爸爸便把孩子带到医院看眼科,不过,医生说孩子太小了,无法测视力,让他们再等等。过了一年再测时,孩子已是2000多度的近视,而这个年龄的孩子正常应该有300度的远视储备。孩子爸爸又把孩子带到另一家医院就诊,检查发现孩子晶状体异常。
近半年来,家人又发现,孩子走路姿势与其他同龄孩子不一样,略微驼背。一家人又带着孩子到广东深圳一家医院的骨科就诊,经脊柱X光拍片发现腰椎稍后凸。
除此之外,孩子一直食欲不好,个子明显高于同龄男孩,但却十分消瘦。到一家医院的儿保科就诊后,孩子被诊断为“营养不良,生长过快”。
就这样反复求医,孩子的状况不见好转。也不知道具体原因。
又高又瘦手指脚趾偏长
家属被迫四处求医,最终被发现是一种罕见病——马凡综合征。
马凡综合征
马凡综合征是常染色体显性遗传病,由法国儿科医生马凡(Marfan)在1896年首次报道。马凡综合征是遗传性结缔组织病,以骨骼、眼及心血管三大系统的病变为主要特征。
马凡综合征(Marfan syndrome)患者身材高大、四肢修长,因为身形特点在一些领域极有优势,所以也被称为“天才病”。美国的女排名将海曼也是马凡综合征患者,她身材高瘦,手指特长,叱咤排坛多年。不过她33岁那年猝死在球场上,也是因为马凡综合征。
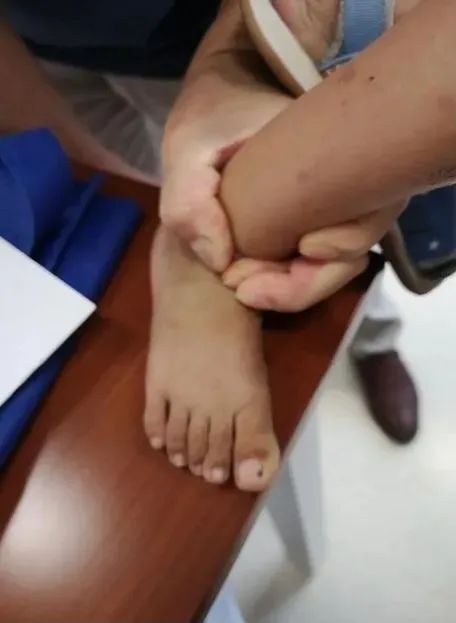
马凡综合征还有一个表现是身材异常高大。一测量,孩子的身高达96cm,超过同龄男孩的90多个百分位。但他的体重只有12.4公斤,接近同龄孩子对照体重的下限。再看看手指及脚趾,都较长。这更加符合马凡综合征的特征,这种病又被称为“蜘蛛指(趾)”症,原因就是患者的手指和脚趾都偏长,让人联想到蜘蛛。
因马凡综合征常常累及心脏,仔细听诊,心前区没有杂音,马上作心脏超声,结果发现“主动脉窦部增宽”。马凡综合征患者三大系统的病变特征,孩子都有了。
经询问,孩子父母表示自己及家族均没有患这种病。
最终基因检测发现,检出FBN1 基因上与受检者表型部分相关的2个疑似致病变异,结合临床表现,可以确诊为马凡综合征。
该病与普通遗传病一样,目前还没有根治的办法,只能对症处理。
“天才病”患病率低于万分之一
马方综合征又称为马凡综合征,为一种遗传性结缔组织疾病,为常染色体显性遗传,患病特征为四肢、手指、脚趾细长不匀称,身高明显超出常人,伴有心血管系统异常,特别是合并的心脏瓣膜异常和主动脉瘤。该病同时可能影响其他器官,包括肺、眼、硬脊膜、硬颚等。马凡综合征的发病率约0.04‰~0.1‰。流行病学统计显示,大多数马凡综合征患者有家族史,但同时约有15%-30%的患者是由于自身变异导致的。这种自身突变率大约是1/20000。

马凡综合征的临床表现有骨骼肌肉系统、眼和心血管系统等几方面的异常,如四肢细长、蜘蛛指(趾)、漏斗胸、鸡胸、脊柱后凸、脊柱侧凸;晶体状脱位或半脱位、高度近视、白内障、视网膜剥离、虹膜震颤;约80%的患者伴有先天性心血管畸形。马凡综合征患者日常应避免剧烈运动、过于劳累。
主要临床表现如下:
1.骨骼肌肉系统
主要有四肢细长,蜘蛛指(趾),双臂平伸指距大于身长,双手下垂过膝,上半身比下半身长。长头畸形、面窄、高腭弓、耳大且低位。皮下脂肪少,肌肉不发达,胸、腹、臂皮肤皱纹。肌张力低,呈无力型体质。韧带、肌腱及关节囊伸长、松弛,关节过度伸展。有时见漏斗胸、鸡胸、脊柱后凸、脊柱侧凸、脊椎裂等。
2.眼
主要有晶状体脱位或半脱位、高度近视、白内障、视网膜剥离、虹膜震颤等。男性多于女性。
3.心血管系统
约80%的患者伴有先天性心血管畸形。常见主动脉进行性扩张、主动脉瓣关闭不全,由于主动脉中层囊样坏死而引起的主动脉窦瘤、夹层动脉瘤及破裂。二尖瓣脱垂、二尖瓣关闭不全、三尖瓣关闭不全亦属本征重要表现。可合并先天性房间隔缺损、室间隔缺损、法乐四联症、动脉导管未闭、主动脉缩窄等。也可合并各种心律失常如传导阻滞、预激综合征、房颤、房扑等。
诊断
马凡综合征的主要危害是心血管病变,特别是合并的主动脉瘤,应早期发现,早期治疗。根据临床表现骨骼、眼、心血管改变三主征和家族史即可诊断。临床上分为两型:三主征俱全者称完全型;仅二项者称不完全型。诊断此病的最简单手段是超声心动图,有怀疑者均可行此检查,进一步确诊则需要通过MRI(磁共振显像)。
现代的诊断标准是在1986年第七届人类遗传学国际大会上建立,并在1988年第一次国际马凡综合征专题讨论会上明确规定的标准,1996年,de Paepe A 重新修正被视为统一标准。马凡氏综合征的主要危害是心血管病变,特别是合并的主动脉瘤,应早期发现,早期治疗。根据临床表现骨骼、眼、心血管改变三主征和家族史即可诊断。
临床上分为两型:三主征俱全者称完全型;仅二项者称不完全型。诊断此病的最简单手段是超声心动图,有怀疑者均可行此检查,进一步确诊则需要通过MRI(磁共振显像)。
马凡综合征一般表现为连续传递遗传或隔代遗传。马凡氏综合征患者的家属也应提高警惕,可做全面的身体检查,及时诊断是否患病
治疗
1.一般治疗
目前尚无特效治疗。有人主张应用男性激素及维生素,对胶原的形成和生长可能有利。对先天性心血管病变宜早期手术修复,对心功能不全、心律失常者宜内科治疗。一旦确诊为合并有主动脉瘤或心脏瓣膜关闭不全,则应视情况考虑手术治疗,因为药物是不能去除此病的。由于动脉瘤有破裂出血的危险,心脏瓣膜关闭不全也有致心衰死亡的危险,所以尽管手术有一定风险,专家们还是建议手术治疗。事实上,随着科技进步,目前手术成功率已在90%以上。若提示有主动脉夹层动脉瘤破裂者,应及时手术治疗。
2.手术治疗
主动脉根部置换术
-
适应证:主动脉根部显著扩张的患者无论症状如何,都应考虑接受手术干预。
-
目的:防止血管破裂。
-
注意:主动脉根部严重扩张之前行择期置换术效果优于明显扩张或夹层时的急诊修复术。
-
儿童和青少年阶段可通过定制的背部支架进行校正;
-
如果侧弯程度太大,可进行外科矫正手术。
胸骨矫正
通过矫正手术减少胸骨畸形造成的压迫,改善心肺功能。
眼科手术
-
视网膜脱落:进行手术修复;
-
白内障:人工晶状体代替。
预后
病变发展速度个体差异很大,但总体看预后险恶。据Mardoch等调查,有1/3的马方综合征患者死于32岁以前,2/3死于50岁左右。1995年SilevermanJL报告平均年龄仅40岁。死亡的主要原因绝大多数是心血管病变造成的。最常见的是主动脉瘤破裂、心包压塞或主动脉瓣关闭不全和二尖瓣脱垂而致的心力衰竭或心肌缺血。超声心动图检查证明有95%的患者均存在不同程度的心血管病变,Crawford的研究表明,当主动脉根部直径大于6cm时,则有出现主动脉瓣关闭不全和发生主动脉内膜剥离症的危险。
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言






目前罕见病的普及比较少,这种通过典型症状和超声检查便可以初步诊断的疾病,建议基层医务人员了解,早发现,早干预,帮助患儿纠正不良姿势、建立合适的运动习惯,有远期的正面影响。
97
孕中做个单基因筛查就好了,就不会导致家长这么迷茫到处求医。还望单基因筛查能够在孕中人群中能够普查。
102
儿童眼病筛查确实很有必要
107
太可怕了吧
127