阿尔茨海默病中的小胶和星胶通过补体C1q依赖途径协调清除突触
2022-11-17 brainnew神内神外 brainnew神内神外
慢性神经炎症,表现为胶质细胞异常、促炎细胞因子水平升高和突触丢失,是AD的主要病理特征之一。在AD小鼠模型和AD病人大脑中异常过度激活导致神经元损伤和突触丢失的一种途径是经典补体途径(CCP)。
慢性神经炎症,表现为胶质细胞异常、促炎细胞因子水平升高和突触丢失,是AD的主要病理特征之一。在AD小鼠模型和AD病人大脑中异常过度激活导致神经元损伤和突触丢失的一种途径是经典补体途径(CCP)。AD患者的大脑和脑脊液 (CSF) 中CCP因子异常升高。人类遗传关联研究也支持该补体途径参与AD的发病机理。补体通路的失调可能在多种中枢神经系统疾病中发挥作用。
为了支持补体的神经毒性作用,在AD、MS、额颞叶痴呆和神经侵入性病毒感染的小鼠模型中,对补体途径的药理或遗传抑制可改善神经退行性变和突触丢失。在发育过程中小胶质细胞通过吞噬多余的突触来改善神经元回路。除小胶质细胞外,在成人脑和疾病中,星形胶质细胞也显示在发育过程中清除突触。与小胶质细胞相反,星形胶质细胞吞噬突触在生理条件下似乎不依赖于c1q。虽然反应性星形胶质细胞在不同的中枢神经系统疾病 (包括AD、HD、PD和MS) 中具有突触毒性作用,但其分子机制在很大程度上仍不清楚。
近期,《Nature Aging》期刊发表了题为“Complement C1q-dependent excitatory and inhibitory synapse elimination by astrocytes and microglia in Alzheimer's disease mouse models”的研究论文,作者团队通过整合的多组学分析发现在AD小鼠模型中星形胶质细胞及小胶质细胞通过Cq1补体途径接触及清除突触,C1q缺失在TauP301S转基因小鼠中具有神经保护作用,提示抑制补体是改善AD等神经退行性病变的有吸引力的策略。
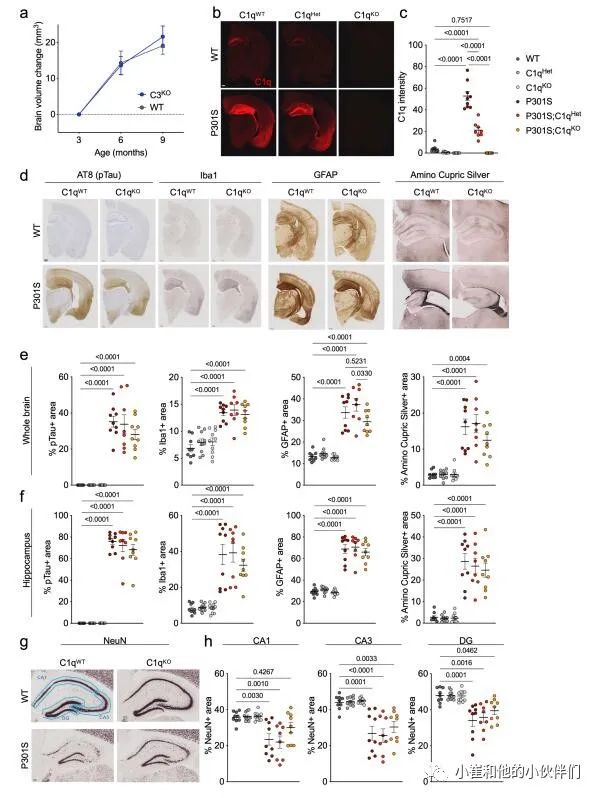
 C1q缺失减少P301S小鼠的神经变性
C1q缺失减少P301S小鼠的神经变性
为了研究C1q在P301S小鼠神经退行性变中的作用,作者团队对C1q进行了基因消融,对雄性小鼠使用磁共振成像 (MRI),行为学和病理分析以及转录组学和突触蛋白质组学分析(图1a)。
与野生型(WT)小鼠相比,C1q缺陷(C1qKO)小鼠在成熟过程中的脑容量增长率以基因剂量依赖性的方式降低(图1b,c),但没有观察到C3KO小鼠脑容量变化的差异(补充图1a),表明C1q在大脑中可能具有不依赖CCP的生理功能。在P301S小鼠中,在6到9个月间,脑容量显著减少,反映神经退行性变(图1c,d)。在P301S;C1qKO小鼠中,9月龄时脑容量损失较轻,且脑容量与C1qKO小鼠无显著差异(图1c,d)。海马是受Tau病理和神经胶质细胞影响最严重的大脑区域,作者观察到P301S小鼠即使在6个月时海马体积也会减少,并且在6到9个月之间进一步萎缩,而在P301S;C1qKO小鼠海马体积损失被延迟,在6个月时具有显著的保护作用 (图1e,f)。与纯合的C1q敲除 (C1qKO) 所提供的保护相反,P301S;C1qHet小鼠与P301S小鼠相似,这意味C1q减少大于50%才能预防TauP301S神经变性。
为了评估C1q缺失的行为后果,作者评估了9个月大小鼠在开阔区域的运动活动(图1g)。正如预期的那样P301S小鼠表现出过度活跃,这被认为是由海马损伤引起的。虽然在没有P301S转基因的情况下,C1q基因型对运动能力没有影响,但P301S;C1qKO小鼠的过度活动被挽救,而P301S;C1qHet小鼠没有(图1g)。
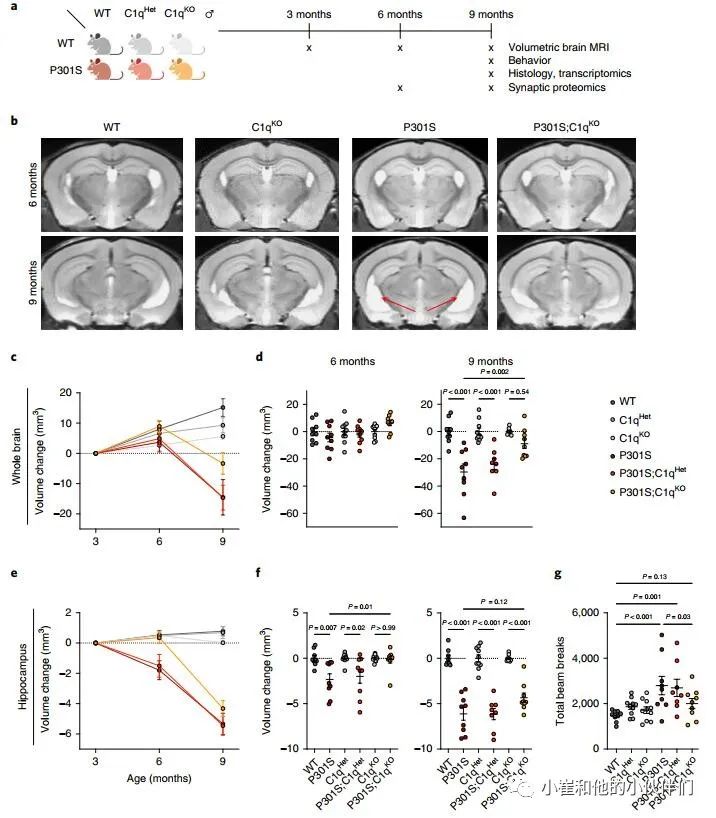
图1 C1q缺失减少P301S小鼠的神经退行性变
接着作者团队分析了9个月大的小鼠的大脑组织病理学。P301S大脑的C1q免疫反应性明显增强,而P301S;C1qHet大脑的C1q免疫反应性降低了约50%,P301S;C1qKO大脑的C1q免疫反应性未检测到(补充图1b,c)。P301S脑的特点是具有强的磷酸化Tau免疫反应性,小胶质细胞和星形胶质细胞分别通过Iba1+和胶质纤维酸性蛋白 (GFAP) 面积增加来测量(补充图1d-f)。与P301S小鼠相比,P301S;C1qHet与P301S小鼠的这些组织病理学读数无差异,P301S;C1qKO与P301S小鼠相比有轻微降低的趋势(补充图1d-f)。
作者观察到氨基铜染色减少的趋势,这反映了P301S;C1qKO海马区受损神经元和神经元标记NeuN密度增加(补充图1e-h)。P301S海马体RNA-seq显示许多基因的上调,包括激活的小胶质细胞和星形胶质细胞的多个标记物(补充图2);然而,C1q缺失对P301S海马的这些主要转录组变化没有影响(补充图2)。总的来说,这些结果表明,C1q缺失减少了P301S大脑退化并使行为正常化,但对Tau病理、神经胶质细胞增生或神经胶质细胞转录变化的程度没有显著影响。因此,C1q缺失的保护作用似乎在Tau病理和总体的神经胶质反应的下游起作用。
 补充图1 5xFAD小鼠脑内Hcar2诱导、APPPS1和PS19小鼠模型中
补充图1 5xFAD小鼠脑内Hcar2诱导、APPPS1和PS19小鼠模型中
Hcar2表达以及Hcar2抗体ab81825的验证
C1qKO减弱P301S小鼠突触中的蛋白质组变化
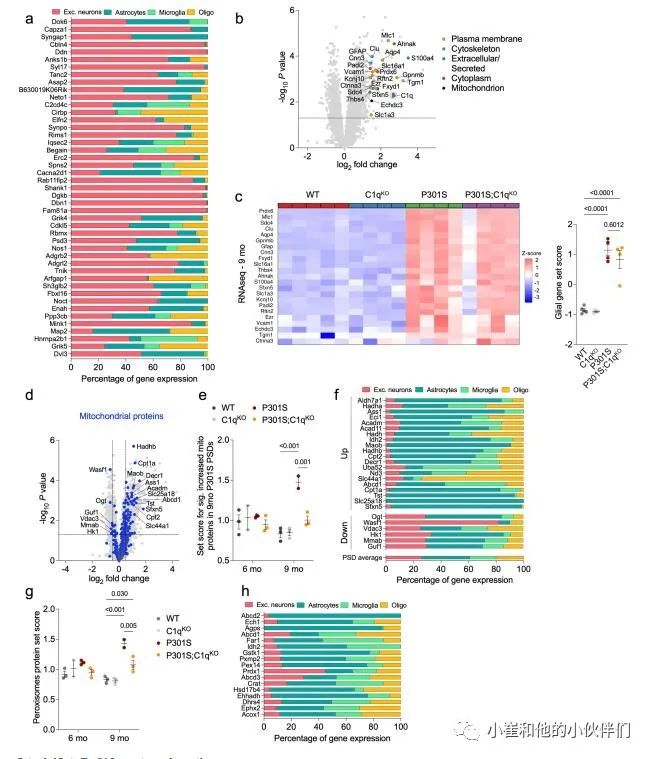
作者团队接下来研究了P301S和C1q基因型突触蛋白组成的变化。作者分别从6个月和9个月大的雄性小鼠中分离出海马突触后密度(PSD)组分,以检测疾病早期和晚期的变化(图2a)。这些部分在PSD和突触后膜的蛋白质中高度富集;它们还包含突触前末端、跨突触粘附分子和一些胶质细胞特异性蛋白(这可能反映了胶质细胞与突触的密切相互作用)的成分。因此,作者团队在整个研究中交替使用术语PSD和突触分数。多重串联质量标签(TMT)蛋白质组学在6个月大的小鼠的PSD片段中共检测到7101个蛋白质,在9个月大的小鼠的PSD片段中检测到4175个蛋白质(图2b和补充表1)。虽然作者之前使用无标签蛋白质组学对9个月大的P301S雌性小鼠的分析中检测到较少的蛋白质,但在当前的研究中也发现了几乎所有的蛋白质(图2b)。
与WT相比,C1qKO小鼠的突触片段在两个年龄均仅显示少量差异表达(DE)蛋白(图2c和补充图3a,c)。在6个月大的P301S突触片段中,作者发现108个下调蛋白和68个上调蛋白(占总蛋白的2.5%;图2 c, e)。9个月时,有253个蛋白下调,434个蛋白上调,占总蛋白的16.5%(图2c,e)。相比之下,P301S;C1qKO突触片段在6个月时显示只有17个DE蛋白减少和19个DE蛋白增加(占总蛋白的0.5%),而在9个月时79个DE蛋白减少和224个DE蛋白增加(占总蛋白的7%)(图2c,f)。在比较P301S;C1qKO和P301S 9个月时的突触时,作者一致发现了许多DE蛋白(图2c和补充图3b),并且随着C1q缺失,Tau依赖性变化减少(补充图3d)。C1q缺乏不影响P301S大脑突触部分的Tau水平(图2e,f)。因此,C1q缺失减弱了Tau病理诱导的年龄依赖性变化,尽管C1q缺失在非转基因小鼠中几乎没有影响(图2d和补充图3d)。由于C1q的缺失并没有显著改变P301S大脑的转录组变化(补充图2),突触蛋白质组的变化很可能是由突触的局部蛋白质变化引起的。
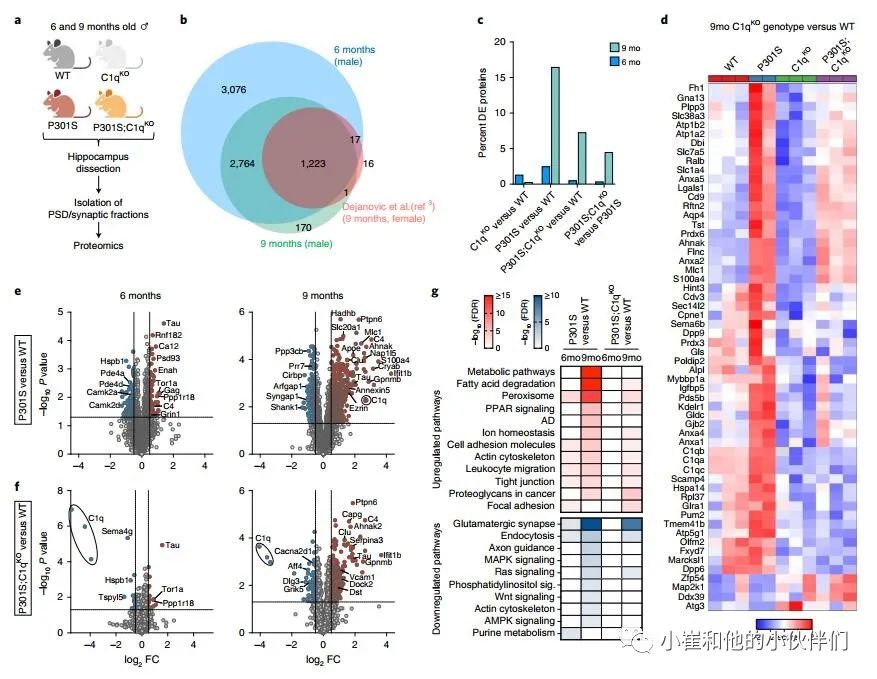 图2 C1q缺失减弱P301S突触的蛋白质组变化
图2 C1q缺失减弱P301S突触的蛋白质组变化
作者评估了P301S转基因和C1q缺失对突触蛋白组中不同功能类蛋白质的影响(补充图3e)。与WT突触相比,P301S中许多核心PSD蛋白,如谷氨酸受体、支架蛋白、突触粘附分子和某些突触前活性区蛋白在6个月时趋于增加,但在9个月时减少(补充图3e)。这可能反映了疾病早期的代偿性突触变化,在后期被突触损伤所克服。利用突触基因本体论工具SynGO,作者团队证实了证实了9个月大的P301S PSDs中下调的DE蛋白显著富含突触组织和典型的突触前和突触后蛋白(补充图3f)。虽然P301S;C1qKO突触片段的突触功能在质量上相似,但其变化不如P301S PSDs显著(补充图3f)。
对P301S突触DE蛋白的KEGG途径分析显示,“代谢途径”(主要包含线粒体蛋白如Echs1、Maob和Atp8)、“脂肪酸降解”(如Adh5、Hadha和Hadh)、“过氧化物酶体”(如Dhrs4、Ehhadh和Abcd1)、“过氧化物酶体增殖物激活受体(PPAR)”信号(如Cpt1a和Cpt2)显著富集,“阿尔茨海默病”(例如Adam10和APOE)和离子稳态(例如Slc4a4和Aqp4)在9个月时尤其突出(图2g)。在P301S;C1qKO突触中,这些通路的增加相对较弱,在6个月时没有明显增加,9个月时的变化类似于6个月时P301S PSDs的变化(图2g)。与此同时,与P301S相比较,在9个月大的P301S突触中增加的大多数通路在P301S;C1qKO中减少(补充图3b,c)。值得注意的是,“代谢途径” 是9个月大的P301S突触中最显着增加的途径,在P301S;C1qKO中没有显著诱导(图2g)。我们还注意到,膜联蛋白是9个月大的TauP301S突触中诱导程度最高的蛋白质之一,并在P301S;C1qKO中被部分归一化(图2d和补充图3d)。
对P301S突触中DE蛋白减少的分析突出了包括“谷氨酸突触”(如Shank1和Grin2a)、“内吞作用”(例如Vps4a和Rab11Fip2)、“轴突引导”(如Ntng1和Smad2)、“MAPK-”(如Mapk1和Map4k4)、“Ras-”(如Ksr1和Syngap1)、“磷脂酰肌醇-”(如Dgkb和Dgkq)和“wnt -信号”(Apc2和Dvl3)和“ampk -信号”(Ppp2r5c和Prkaa2)等通路(图2g)。在6个月大的P301S;C1qKO突触中,没有通路显著减少,仅“谷氨酸能突触”、“胞吞”和“ras信号”在9个月时显著减少 (图2g)。在P301S PSD片段中,“肌动蛋白细胞骨架”通路的不同蛋白显著增加(如ezrin和gelsolin)或减少(如Baiap2和Pak6)。在突触部分中,至少有一些增加的肌动蛋白调节蛋白是由可能与突触相关的胶质细胞表达的。
由于突触蛋白突变导致多种神经和神经精神疾病,作者团队研究了DE蛋白在相关性状和疾病的全基因组关联研究(GW As)中是否富集了遗传信号。对于P301S和WT突触中上调和下调最多的1000个蛋白质,作者使用分层连锁不平衡(LD)评分回归(方法)测试了752个性状的多基因信号,这些性状主要来自英国生物银行(UKBiobank),并选择GWAS研究。作者在将性状归类为23个类别或域后,发现9个月大的P301S和P301S;C1qKO 突触中下调的蛋白在认知、精神和活动域有显著的丰富,包括教育成就、智力得分、认知表现和概念插值(补充图4a)。相比之下,9个月大的P301S PSD片段中上调的蛋白或6个月大的DE蛋白中这些性状的富集有限(补充图4b)。这表明9个月大的P301S和P301S;C1qKO突触蛋白下调与人类认知功能和行为有关。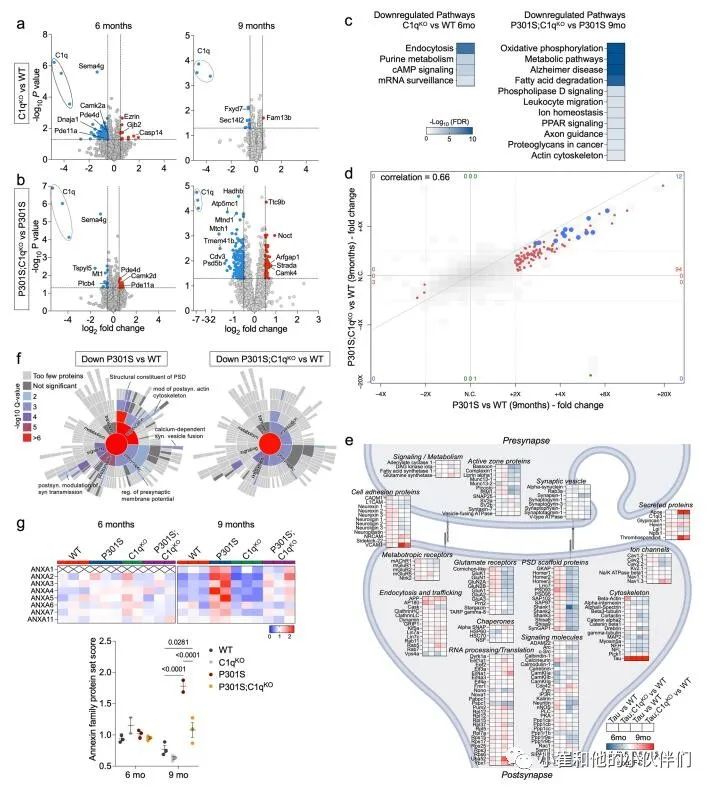 补充图3 P301S小鼠C1q依赖性突触蛋白质组的变化
补充图3 P301S小鼠C1q依赖性突触蛋白质组的变化
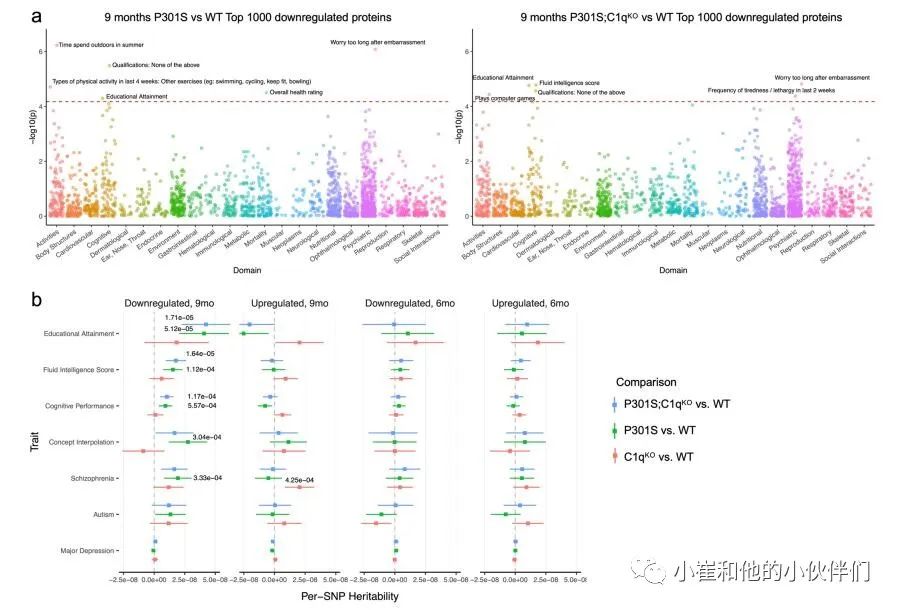 补充图4 P301S和P301S;C1qKO突触部分鉴定的差异丰富蛋白质的遗传富集分析
补充图4 P301S和P301S;C1qKO突触部分鉴定的差异丰富蛋白质的遗传富集分析
P301S突触处胶质细胞蛋白的C1q依赖性升高
作者注意到许多典型的星形细胞特异性蛋白,如Aqp4、Mlc1和Slc1a4在9个月大的P301S突触片段中以c1q依赖的方式增加(图2d)。虽然星形细胞蛋白的污染是可能的,但作者也考虑了星形胶质细胞蛋白与突触制剂的共聚是否引起星形胶质细胞与突触的密切相互作用。作者从P301S海马中生成伪体单细胞RNA测序(scRNA-seq)数据,发现55个高度上调蛋白中的大多数主要由神经胶质细胞表达,而不是兴奋性神经元(图3a)。
相比之下,减少最多的蛋白质主要由兴奋性神经元产生(补充图5a)。除Aqp4和Mlc1外,许多其他上调的DE蛋白选择性或主要由星形胶质细胞表达(例如,簇蛋白、Slc1a3、Sdc4、AHNAK、ezrin、GFAP和Thbs4)(图3a)。少量上调蛋白主要由小胶质细胞表达(例如,Gpnmb和Myo1f)(图3a)。作者假设,9个月大的P301S突触部分中胶质蛋白的激增可能反映了其分泌和随后在突触处的积累的增加,和/或胶质突起与受损突触接触的增加。一致地,许多增加的胶质蛋白要么定位于质膜(例如Slc16a1和Aqp4)、细胞骨架(例如ezrin),要么是细胞外/分泌的(clusterin和Thbs4;补充图5b),并且已知存在于星形胶质细胞过程中。
值得注意的是,P301S突触部分中胶质蛋白的增加是C1q依赖性的(图3b)。这反对用胶质衍生蛋白不加区分地污染PSD制剂。P301S;C1qKO突触中胶质蛋白的相对减少似乎是由于胶质蛋白与突触部分的结合发生了特定变化,而不是胶质蛋白的总体丰度,因为P301S与P301S;C1qKO大脑中相应基因的表达没有显著差异(补充图5c)。
在P301S突触中增加最多的蛋白质是星形胶质细胞特异性线粒体蛋白Echdc3和Sfxn5 (图3a,b)。许多线粒体蛋白在P301S突触片段中以年龄和C1q依赖的方式增加(补充图5d,e)。中枢神经系统细胞类型之间的能量代谢不同,在9个月时,P301S中的“代谢途径”显著升高,但P301S;C1qKO突触没有升高(图2g)。
尽管线粒体可以在病理条件下从星形胶质细胞转移到神经元,但作者推断,突触部分的线粒体可能来自与突触有密切物理接触的神经胶质过程。一直以来,P301S突触中增加最多的20个线粒体蛋白主要由星形胶质细胞表达(例如,Maob, Cpt1a和Tst, Slc25a18),而显著减少的线粒体蛋白有广泛的表达模式,包括更强的神经元产生(例如,Wasf1)(补充图5f)。同样,在9个月大的P301S突触片段中以C1q依赖性方式增加的过氧化物酶体蛋白也主要由星形胶质细胞表达(补充图5g)。
 补充图5 突触胶质蛋白的丰度及其相应基因的表达
补充图5 突触胶质蛋白的丰度及其相应基因的表达
接下来,作者团队使用免疫电子显微镜(IEM)来确定P301S小鼠中星形胶质细胞与突触的物理联系是否发生改变。他们通过星形胶质细胞特异性谷氨酸转运蛋白EAAT2/Glt1的免疫标记鉴定了星形胶质细胞过程,并量化了与海马齿状回(DG)和CA1区突触接触的星形胶质细胞进程的长度(图3c和补充图6a)。与WT小鼠相比,P301S小鼠的DG和CA1区与突触相关的星形细胞突起的长度增加了约两倍(图3d和补充图6b)。P301S小鼠的平均突触周长没有变化,这意味着星形胶质细胞接触了更大比例的突触膜(图3e和补充图6c)。值得注意的是,星形胶质细胞-突触关联的程度与C1q标记的突触前百分比显著相关3(图3f)。
作者使用共焦显微镜定量了海马CA1区中兴奋性突触(Homer1 puncta)与GFAP+星形胶质细胞表面的空间接触(图3g)。由于丢失一个C1q拷贝对P301S小鼠的病理学没有影响(图1和补充图1),作者在该分析中将P301S和P301S;C1qHet大脑分组,以增加统计能力。虽然C1qKO与WT海马之间没有差异,但我们观察到P301S海马的表面GFAP–Homer1关联显著增加,而P301S;C1qKO与P301S相比显著降低(图3h)。使用细胞质星形胶质细胞标记蛋白S100b来显示星形胶质细胞体积,证实了P301S小鼠中星形胶质细胞–Homer1关联的C1q依赖性增加(补充图6d,e)。总之,作者对突触蛋白质组学数据、IEM和免疫组织化学(IHC)测量的分析表明在突触吞噬和丢失的疾病阶段,星形胶质细胞可以以C1q依赖的方式增加与突触的相互作用。
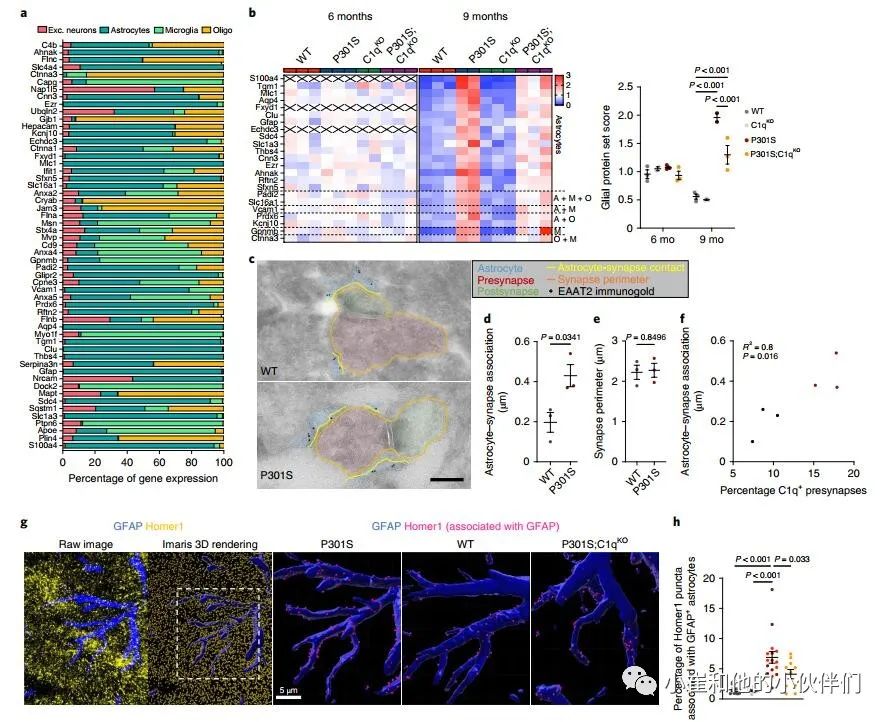 图3 胶质蛋白在P301S突触处升高,并通过C1qKO标准化
图3 胶质蛋白在P301S突触处升高,并通过C1qKO标准化
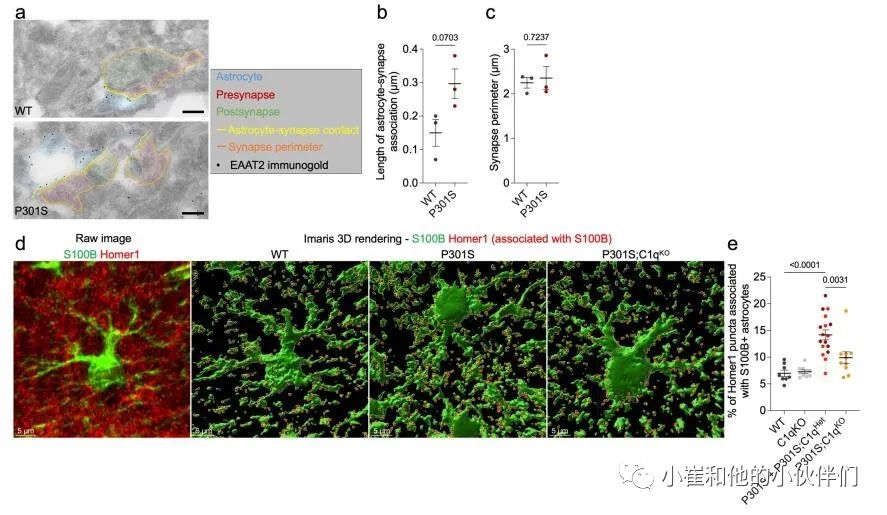 补充图6 星形胶质细胞-突触相互作用的iEM和iHC分析
补充图6 星形胶质细胞-突触相互作用的iEM和iHC分析
AD大脑突触中的胶质蛋白升高
作者想知道AD患者突触部分的胶质蛋白是否也升高了。与最近发表的AD患者颞上回(BA 41/42)突触神经元蛋白质组数据的比较显示,人类AD与对照组的变化和9个月大的P301S与WT小鼠突触蛋白质组之间存在显著的正相关(图4a)。值得注意的是,在P301S突触片段中升高的胶质蛋白,包括补体因子C1q和C4、星形细胞标记蛋白MLC1和GFAP、小胶质GPNMB和AHNAK以及膜联蛋白是AD突触小体中升高最多的蛋白(图4a)。
作者推断在患者的CSF中可以检测到C4水平升高和CCP的激活,这可能是补体激活的一个有用的生物标志物。在AD患者的CSF中,总C4浓度和和加工 (裂解和活化) C4浓度显著增加 (图4b)。在来自独立患者队列的CSF中也观察到类似的结果,其趋势是总C4升高,而加工的C4显著增加。相比之下,补体因子B (替代补体途径的组成部分) 在AD CSF中没有明显改变 (补充图7)。相比之下,补体因子B (替代补体途径的组成部分) 在AD CSF中没有明显改变 (图4b和补充图7)。激活的Bb亚单位水平在对照组和AD脑脊液中非常低,但在AD脑脊液中确实显示出升高的趋势(图4 B和补充图7)。因此,P301S突触蛋白组中胶质蛋白的上调也出现在AD中,可能与疾病的病理生理有关。AD患者脑脊液中C4(和C3)水平升高与CCP在阿尔茨海默病神经退行性变中的作用一致。
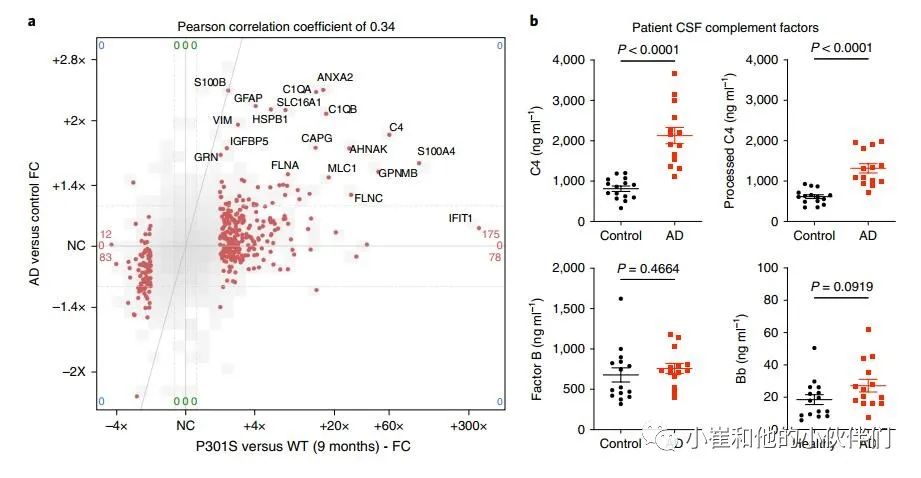
图4 人AD突触部分的胶质蛋白增加,AD CSF中的C4升高
P301S小鼠的胶质C1q依赖性突触消除
根据作者团队的蛋白质组学、IEM和IHC数据,提出星形胶质细胞可能在突触吞噬过程中以C1q依赖的方式与突触相互作用的假设。为了分析星形胶质细胞和小胶质细胞吞噬突触,作者团队对GFAP+星形胶质细胞、Iba1+小胶质细胞、Lamp1+溶酶体以及兴奋性突触后标记Homer1进行免疫染色(图5a)。由于抑制性突触在AD中也受到影响,我们还对抑制性突触后标记物gephyrin进行了免疫标记。通过共聚焦显微镜和海马CA1区域的3D重建,在同一幅图像中测量了小胶质细胞和星形细胞溶酶体内的Homer1和gephyrin斑点的数量(图5a)。
值得注意的是,使用GFAP或S100B,作者基本上识别了相同的星形细胞Lamp1+结构群体(补充图8a)。正如之前的研究所预期的P301S海马中的小胶质溶酶体包含兴奋性突触,与WT对照相比,Homer1点增加了约10倍(图5b)。与P301S相比,P301S;C1qKO大脑中Homer1的小胶质吞噬能力明显降低(图5b)。值得注意的是,作者还在星形细胞溶酶体中发现了相当一部分Homer1斑点,在P301S海马区增加了5到10倍(图5c)。P301S;C1qKO脑星形细胞溶酶体内Homer1点的比例显著降低,表明P301S小鼠兴奋性结构的星形细胞吞噬至少部分依赖于C1q(图5c)。小胶质细胞和星形胶质细胞溶酶体中也存在gephyrin 斑点 (图5d,e)。与Homer1一样,在P301S海马中,小胶质细胞和星形胶质细胞对gephyrin的摄入升高,并且部分依赖于C1q(图5d,e)。然而,在健康大脑中,兴奋性和抑制性突触的吞噬不受C1q丢失的影响,因为WT和C1qKO海马在胶质溶酶体中Homer1和gephyrin的含量同样低(图5b-e)。吞噬突触点的数量与跨基因型星形细胞溶酶体和小胶质溶酶体的体积变化相对应(补充图8b)。
值得注意的是,在所有基因型中,始终在星形细胞内发现了更多的Homer1点,而不是小胶质溶酶体 (图5f)。相反,在P301S海马的星形细胞溶酶体和小胶质细胞溶酶体中,gephyrin puncta含量较低 (不考虑C1q基因型),与非转基因WT动物的星形胶质细胞和小胶质细胞中gephyrin puncta含量相似 (图5g)。在P301S大脑中,这种吞噬gephyrin puncta的小胶质细胞倾向尤其明显,在小胶质细胞中经常观察到强烈的gephyrin puncta免疫反应性积累,而不是星形细胞溶酶体(补充图8c)。一种可能性是,这种强烈的免疫反应可能反映了含有许多抑制突触的树突片段的去除。与P301S;C1qKO大脑中突触吞噬减少一致,在C1q缺陷的P301S小鼠中,兴奋性和抑制性突触丢失得到改善(图5h)。
最后,作者测试了星形细胞吞噬突触结构是否需要C3, 这是C1q下游的中枢补体成分。P301S小鼠小胶质细胞和星形胶质细胞对Homer1和gephyrin的吞噬显著增加,P301S;C1qKO小鼠中Homer1和gephyrin的吞噬平均部分减少(补充图8d-g)。然而,只有星形胶质细胞溶酶体中的斑斑凝胶藻素减少在P301S;C1qKO与P301S小鼠中达到统计显著性(补充图8g),可能是由于动物间的高度变异性。就像在C1q实验队列中一样,作者团队在他们分析的每个大脑中星形细胞溶酶体中发现了更多的Homer1点,而在C3实验队列中P301S小鼠的小胶质溶酶体中则优先发现了gephyrin puncta(补充图8h,i)。
接下来,作者分析了P301S;C1qKO队列中兴奋性突触的C3标记。与WT相比,P301S海马中C3+ Homer1瘤的百分比显著增加,而P301S;C1qKO与WT海马相当,与P301S大脑相比显著降低(图5i,j)。P301S与P301S;C1qKO大脑中C3点状突起的总数没有显著差异(图5k),表明C1q的缺失特别影响C3在突触的沉积。总的来说,数据表明C3作用于C1q激活的下游,CCP促进星形胶质细胞和小胶质细胞消除突触。
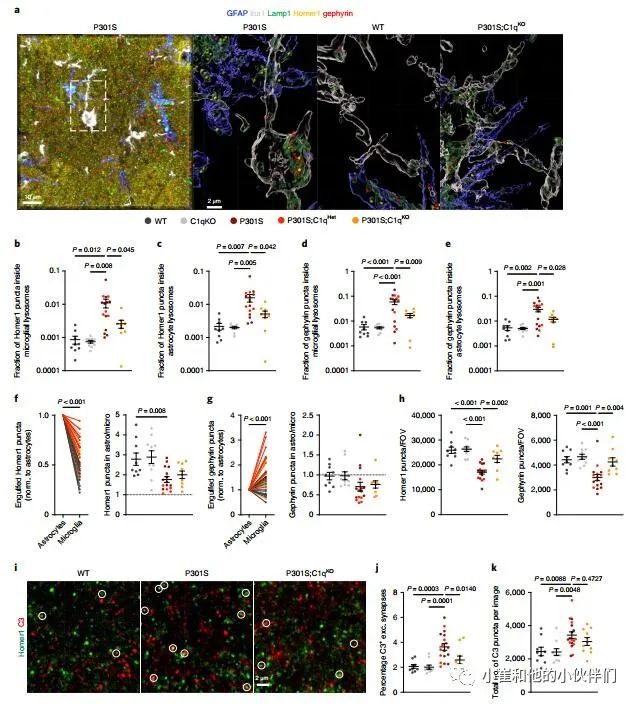 图5 星形胶质细胞和小胶质细胞以C1q依赖的方式消除P301S小鼠兴奋性和抑制性突触
图5 星形胶质细胞和小胶质细胞以C1q依赖的方式消除P301S小鼠兴奋性和抑制性突触
 补充图8 P301S小鼠星形胶质细胞和小胶质细胞对
补充图8 P301S小鼠星形胶质细胞和小胶质细胞对
兴奋性和抑制性突触的补体依赖性吞噬
星形胶质细胞补偿受损的小胶质细胞吞噬功能
小胶质细胞特异性TREM2功能丧失或突变会增加AD风险,而AD小鼠模型中TREM2的丧失对小胶质细胞功能具有深远的影响,并抑制其活化,向Aβ斑块的迁移和吞噬活性。为了检查小胶质细胞功能障碍是否可能影响星形胶质细胞对突触的清除,作者分析了Trem2缺失在AD小鼠模型TauPS2APP(结合了 Aβ淀粉样蛋白和Tau病理)中的影响。在17个月大时,当存在斑块,磷酸Tau,营养不良的轴突和神经胶质增生时,作者对TauPS2APP和TauPS2APP;Trem2KO脑切片进行了免疫染色,用先前建立的方案和对有和无淀粉样斑块的海马CA1区成像(由存在的Lamp1+营养不良轴突识别) (图6a)。在TauPS2APP大脑中,与无斑块区域相比,小胶质细胞和星形胶质细胞在斑块附近吞噬的Homer1和gephyrin puncta更多 (图6b-e)。在TauPS2APP;Trem2KO大脑中,与TauPS2APP小鼠相比,斑块附近小胶质细胞吞噬的突触明显减少 (图6b,c),而星形细胞吞噬Homer1 puncta不受缺乏Trem2的影响 (图6d)。
值得注意的是,与TauPS2APP大脑相比较,在TauPS2APP;Trem2KO中,在斑块附近的星形胶质细胞吞噬gephyrin puncta的作用显着增加(图6e)。与在P301S小鼠中一样,与小胶质细胞溶酶体相比,星形胶质细胞溶酶体含有更多的Homer1 puncta(图6f),而在TauPS2APP小鼠中,gephyrin puncta在小胶质细胞溶酶体中更丰富 (图6g)。TauPS2APP小鼠中trem2缺陷的总体影响是星形细胞和小胶质溶酶体中Homer1和gephyrin的比例增加(图6f,g)。因此,Trem2对于斑块附近的小胶质细胞有效吞噬突触是必要的,星形胶质细胞至少在一定程度上可以补偿抑制突触的小胶质细胞吞噬受损。
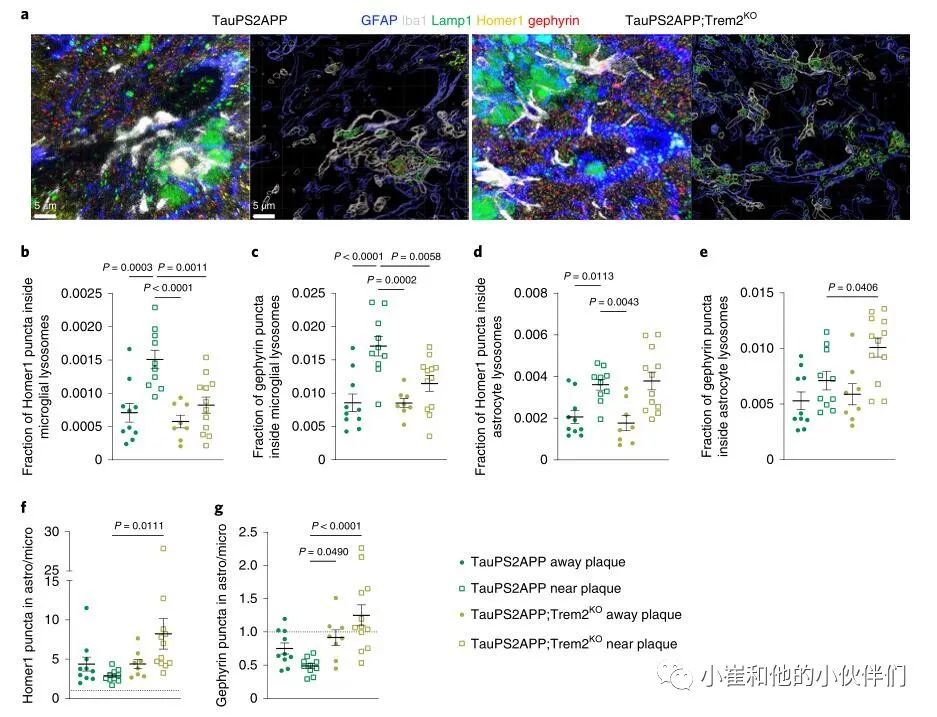 图6 星形胶质细胞补偿Trem2缺陷TauPS2APP
图6 星形胶质细胞补偿Trem2缺陷TauPS2APP
小鼠抑制性突触的小胶质细胞吞噬功能受损
作者团队通过对蛋白质组数据的深度剖析发现星形胶质细胞及小胶质细胞通过补体依赖性方式对兴奋性和抑制性突触清除的新作用。表明在病理生理过程中,星形胶质细胞和小胶质细胞在突触吞噬中的伴随/协调作用。本研究还发现星形胶质细胞和小胶质细胞可能在突触处感知不同的补体分子。总的来说,作者团队的发现促进了对补体介导的突触消除和神经元损伤的潜在机制的理解,确定了小胶质细胞和星形胶质细胞的抑制性突触与兴奋性突触吞噬作用的意外分工,并为AD的潜在治疗方法开辟了新的途径。
作者:brainnew神内神外
版权声明:
本网站所有注明“来源:梅斯医学”或“来源:MedSci原创”的文字、图片和音视频资料,版权均属于梅斯医学所有。非经授权,任何媒体、网站或个人不得转载,授权转载时须注明“来源:梅斯医学”。其它来源的文章系转载文章,本网所有转载文章系出于传递更多信息之目的,转载内容不代表本站立场。不希望被转载的媒体或个人可与我们联系,我们将立即进行删除处理。
在此留言